王磊教授:重视先天性肝纤维化的诊断和治疗
—— 作者: 时间:2021-07-30
阅读数:
73
山东大学第二医院肝病科 王岩 王磊
先天性肝纤维化(congenital hepatic fibrosis,CHF)是1961年由Kerr等首先命名的一种常染色体隐性遗传性疾病,是一种罕见的、与胆管板畸形相关的肝内胆管遗传发育障碍疾病[1]。CHF是引起非肝硬化性门静脉高压疾病的一种,临床表现与肝硬化有相似之处,需肝穿活检明确诊断,在临床中常被漏诊和误诊。
流行病学
CHF发病无性别和地域差异,多呈散发性,可有家族史但少见。有学者报道,2.70%(3/111例)患者有CHF家族史。其发病率极低,有文献报道在1/4万~1/2万,随着对该病的认识和重视,其发病率可能有所增高。任何年龄段均可起病,多于青少年、青年期确诊[2,3]。
发病机制
CHF是一种由多囊肾/多囊肝病变1(polycystic kidney and hepatic disease 1, PKHD1) 基因突变引起的遗传性胆管病变,目前已报道300余种PKHD1基因突变位点及组合[4]。该基因定位于人染色体6p21,最长开放阅读框架约12.2kb,由66个外显子组成,编码纤维囊肿蛋白/多管蛋白(fibrocystin/polyductin,FPC)。此蛋白主要在胆管和肾小管上皮细胞的初级纤毛上表达,在肝脏的生理功能主要为调节胆管上皮分泌胆汁,还可通过促进细胞分化相关蛋白的表达,参与胆管分化成熟的过程[5,6]。PKHD1基因突变会导致胆管细胞的FPC蛋白功能缺陷,胆管板发育畸形。胆管板畸形是CHF基本病理变化,引起未发育成熟的胆管出现进行性、非特异性炎性坏死过程,此过程会募集巨噬细胞等,引起胶原纤维过度沉积,门静脉周围逐渐纤维化(见图1)。
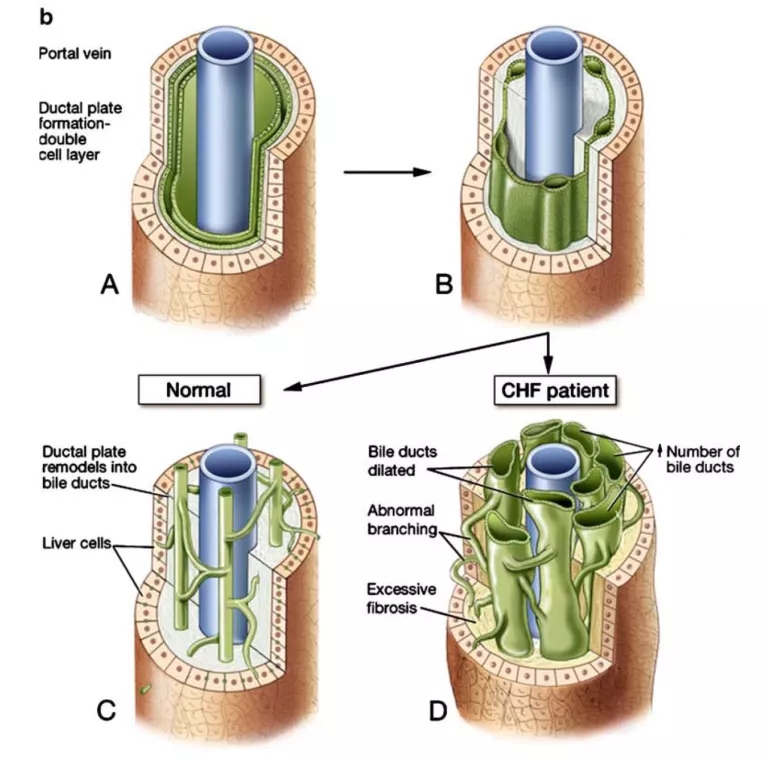
图1.胆管板发育畸形的病理变化
胆管系统的发育起源于门静脉周围的双向潜能肝母细胞,在妊娠期的第4~8周,原始肝母细胞像袖套一样包绕门静脉形成圆柱形单层胆管板,随后发育形成双层胆管板,双层胆管板最终被重塑形成成熟的胆管,未分化的胆管板形成间质[7]。在这个过程中,较大的胆管先发育,至出生后4周最小的胆管才发育完成。在胆管发育的不同阶段出现停滞或异常均会引起胆管板畸形,形成不同的纤维囊性肝脏疾病(见图2)。早期发育异常会引起累及大胆管的疾病(caroli病和胆总管囊肿),中期发育异常会引起累及中等胆管的疾病(多囊肝),晚期发育异常会引起累及小胆管的疾病(CHF和胆管错构瘤)[8]。
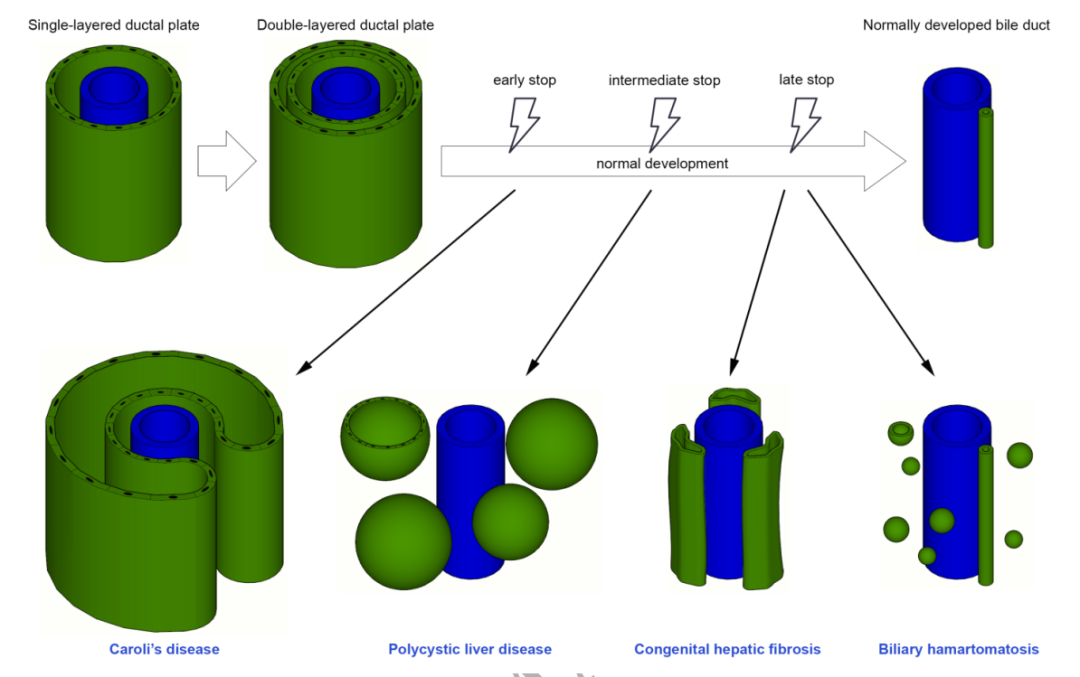
图2.胆管发育停滞或异常所致不同的纤维囊性肝脏疾病
临床表现
1、临床分型
根据其临床表现,CHF可分为门静脉高压型、胆管炎型、门静脉高压与胆管炎混合型和隐匿型四种类型[9]。国内有报道,在75例患者中,门静脉高压型38例(50.7%)、胆管炎型4例(5.3%)、混合型30例(40.0%)、隐匿型3例(4.0%)。
门静脉高压型为主要类型,表现为上消化道出血、腹水、脾大和脾功能亢进等;胆管炎型通常伴发caroli病,表现为间断发热、腹痛等胆管炎的表现;门静脉高压与胆管炎混合型兼有门静脉高压和胆管炎两型特征;而隐匿型无门静脉高压和胆管炎等相关临床表现,需经肝穿刺病理检查才能诊断。门静脉高压型患者可出现WBC和PLT下降,胆管炎型和混合型患者可出现ALP和GGT升高,但所有类型患者白蛋白、凝血指标和胆碱酯酶等反映肝脏合成功能的指标大致正常。这一特点可区分CHF与其他肝损伤导致的肝硬化。
2、伴发疾病
文献报道约60%CHF伴发常染色体隐性遗传性多囊肾病(autosomal recessive polycystic kidney disease,ARPKD),约20%CHF伴发Caroli病,这三种疾病均由于PKHD1基因突变所致的初级纤毛蛋白FPC功能缺陷所导致[10,11]。有国外文献报道CHF还可伴随多种综合征,例如朱伯特综合征、眼-肾综合征、眼-脑-肝-肾综合征、耳蜗前庭综合征等[12],国内尚未见该类报道。
临床诊断
凡遇有不明原因脾大、上消化道出血等门静脉高压表现而肝功能正常或轻度异常的患者,都应考虑到CHF的可能。常用的影像学检查方法包括超声、CT和MRI。CHF患者肝脏影像学改变包括左内叶体积正常或增大、左外叶与尾叶增生和右叶萎缩、脾大和门静脉增宽等[13]。合并Caroli病的CHF患者亦可伴发肝内胆管扩张和肝内胆管结石。合并ARPKD的患者亦可伴发多发肾囊肿。
肝活检是诊断CHF的金标准,其特征性病理特点为:(1)肝小叶汇管区周围弥漫纤维化,中央静脉仍位于肝小叶的中央,肝小叶微循环保持不变,这是与肝硬化假小叶的重要区别;(2)汇管区纤维间隔内可见小胆管增生,内含有许多形态各异的胆管,即胆管板畸形,这是先天性肝纤维化特有的形态;(3)肝细胞板排列大致正常,炎症表现不明显或很轻,一般无肝细胞结节性再生[14]。目前还可通过连锁基因分析和直接检测PKHD1基因的突变来诊断CHF,但由于其基因的复杂性,一般不作为常规检测方法。
治疗和预后
针对CHF患者,目前尚无特效的治疗方法(针对胆管板发育异常)来逆转或停止CHF的进展,临床上主要处理CHF的并发症。食管胃底静脉曲张破裂出血可采用药物、内镜和介入等治疗。TIPS能有效降低门静脉高压,且术后肝性脑病发生率很低。肝移植是目前最佳的根治性治疗方法。
对于CHF患者,若能早期诊断并干预,在有效控制门静脉高压的情况下,预后通常优于肝硬化导致的食管胃底曲张静脉破裂出血。进行肝移植的患者预后最好。
总之,CHF是一种非肝硬化性门静脉高压疾病,临床表现与肝硬化有相似之处,临床诊断较为困难。需结合临床表现和影像学特征,尤其是存在严重门静脉高压而肝功能良好时,应注意到CHF的可能。活组织病理检查是诊断CHF的金标准。提高对CHF的认知对诊断至关重要的。
参考文献:
[1] Kerr D, Harrison CV, Sherlock S, et al. Congenital hepatic fibrosis. Q J Med.1961,30(117):91-117.
[2] Turkbey B, Ocak l, Daryanani K, et al.Autosornal recessive polycystic kidney disase and congenital hepatic fibrosis (ARPKD/CHF). Pediatr Radiol.2009,39(2):100-111.
[3] Veigel MC, Prescott-Focht J, Rodriguez MG,et al. Fibropolycystic liver disease in children.Pediatr Radiol. 2009, 39(4):317-327.
[4] Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large receptor—like protein.Nat Genet.2002,30(3):259-269.
[5] Ward CJ,Yuan D,Masyuk TV,et al.Cellular and subcellular Iocalization of the ARPKD protein:fibrocystin is expressed on primary cilia. Hum MoI Genet.2003,12(20):2703-2710.
[6] Lazaridis KN, Strazzabosco M, Larusso NF. The cholangiopathies:disorders of biliary epithelia. Gastroenter ology. 2004,127(5):1565-1577.
[7] Desmet VJ.Ludwig symposium on biliary disorders--part I.Pathogenesis of ductaI plate abnormalities. Maye Clin Proc.1998,73(1):80-89
[8] Cannella R, Giambelluca D, Diamarco M,et al.Congenital Cystic Lesions of the Bile Ducts: imaging-based diagnosis. Curr Probl Diagn Radiol. 2020,49(4):285-293.
[9] Arnon R,Rosenberg HK,Suchy FJ. Caroli disease, Caroli syndrome,and congenital hepatic fibrosis. Farmington:Humana Press.2010:331-358.
[10] Poddar U, Thapa BR, Vashishta RK,et al. Congenital hepatic fibrosis in Indian children. J Gastroenterol Hepatol. 1999,14(12):1192-1196.
[11] Rock N,Mclin V. Liver involvement in children with ciliopathies. Clin Res Hepatol Gastroenterol. 2014,38(4):407-414.
[12] Shorbagi A,Bayraktar Y.Experience of a single center with congenital hepatic fibrosis:a review of the Iiterature. World J Gastroenter.2010,16(6):683-690.
[13] Akhan <https://pubmed.ncbi.nlm.nih.gov/?term=Akhan+O&cauthor_id=17164079> O, Karaosmano?lu <https://pubmed.ncbi.nlm.nih.gov/?term=Karaosmano%C4%9Flu+AD&cauthor_id=17164079> AD, Ergen <https://pubmed.ncbi.nlm.nih.gov/?term=Ergen+B&cauthor_id=17164079> B.Imaging findings in congenital hepatic fibrosis. Eur J Radiol.2007, 61(1):18-24.
[14] Dhameja N, Rai V,Singh R, et al. Congenital hepatic fibrosis:Report on two cases and its clinicopathological correlation. Ann Pathol Lab Med.2016,3(2):90 -93.
来源:《国际肝病》编辑部
标签:
新知
肝纤维化
发表评论
全部评论
